

|
|
|||
Default scores were based on the following information for all the parameters. User can modify the scores for default sites and can also |
|||
| add any number of new sites if required in the available text areas in the format shown. Each site should be separated by a space. | |||
| The format should be read as follows:G4580A#5: G to A base change at position 4580 is assigned a default score of 5. | |||
| For any Polymorphism, score =1. The score increases for every parameter and the increment depends on the impact of the parameter. | |||
| Links to OMIM and SWISSPROT provided for all genes alongside. | |||
| Go to Index Page |
|||
| SIFT | Sorting Intolerant From Tolerant | ||
| Given a protein sequence, SIFT will return predictions for what amino acid substitutions will affect protein function | |||
| SIFT is a multistep procedure that: | |||
| (1) searches for and chooses similar sequences | |||
| (2) makes an alignment of these sequences | |||
| (3) calculates scores based on the amino acids appearing at each position in the alignment. | |||
| For more information click -> http://blocks.fhcrc.org/sift/SIFT.html | |||
| PolyPhen | Polymorphism Phenotyping | ||
| This tool predicts possible impact of an amino acid substitution on the structure & function of a human protein using | |||
| straightforward physical and comparative considerations. This prediction is based on straightforward empirical rules� | |||
| which are applied to the sequence, phylogenetic and structural information characterizing the substitution. | |||
| PolyPhen uses empirically derived rules to predict that an nsSNP is | |||
| (1) probably damaging, i.e., it is with high confidence supposed to affect protein function or structure | |||
| (2) possibly damaging, i.e., it is supposed to affect protein function or structure | |||
| (3) benign, most likely lacking any phenotypic effect | |||
| (4) unknown, when in some rare cases, the lack of data do not allow PolyPhen to make a prediction | |||
| For more information click -> http://www.bork.embl-heidelberg.de/PolyPhen/ | |||
| PLHOST | Invariant | 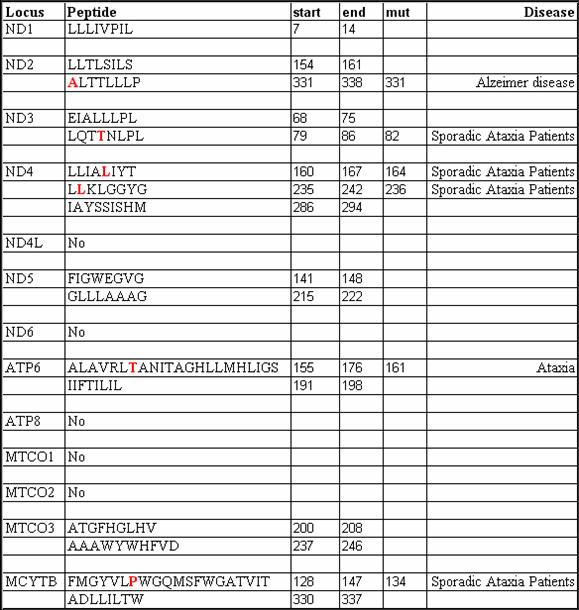
|
|
| peptides | |||
| HRE | The human and rodent mitochondrial genomes contain nucleotide sequences similar to both type of type I & type II HREs | ||
| The steroid and thyroid hormone effects on the mitochondrial genes are direct, concomitant with the effects on nuclear | |||
| genes and involving similar molecular mechanisms to those mediating steroid-thyroid hormone actions on nuclear gene | |||
| transcription (Steroid 61:226-232, 1996). Sequences showing partial similarity to the GRE consensus sequence | |||
| detected in the human mitochondrial genome are as follows: | |||
| 1196������ AGAGGA NNN TGTTCT���� 1210 | |||
| 3218������ AGAACA NNN TTTGTT����� 3232 | |||
| 4103������ TAACCT NNN TGTTCT����� 4117 | |||
| 16494���� CCGACA NNN GGTTCC�� 16508 | |||
| Frequency | Frequency of mutation should be significantly high in patients as compared to normal individuals. If a particular | ||
| of Mutation | mutation appears more often as polymorphism in normal individuals then it should be given a negative score. | ||
| Sites having similar frequencies in both are not informative and hence are not scored. | |||
| HSP & LSP | Heavy Strand Promoter & Light Strand Promoter | ||
| Mammalian mitochondrial genome contains only 2 promoters : LSP & HSP, which produce near-genomic length | |||
| transcripts after that RNA processing release individual mRNAs, tRNAs and rRNAs (Ojala et al. 1981; Clayton, 1991). | |||
| Transcription from LSP is necessary not only for gene expression but also for production of RNA primers required for | |||
| initiation of mtDNA replication (Shadel & Clayton, 1997) [EMBO Journal 2004, 23:4606-4614]. | |||
| POLMRT recognize promoter elements in a sequence-specific manner. Any mutation causing a base change can change | |||
| this interaction effect mtDNA transcription rate. Hence mutations in LSP and HSP are given default high scores. Score | |||
| for LSP is higher because it plays dual role in mtDNA replication and transcription | |||
| 12SRNA | 12 S Ribosomal RNA | ||
| 1095T-C: mutation is to destroy the stem-loop secondary structure, resulting in impaired translation | |||
| (Thyagarajan, D:Ann. Neurology 48::730-736,2000) | |||
| 16SRNA | |||
| Complex I | ND1-6, ND4L | Generate NAD used in the TCA cycle for conversion of Fumarate to OAA and gives lone pair of electron | |
| (from NADH) to the ETC | |||
| Complex I can be subdivided into 3 main fractions: the flavoprotein fragment, the iron-protein fragment, | |||
| and the hydrophobic protein fragment | |||
| Of the 7 mitochondrial DNA Complex I genes, the gene products for MTND1, MTND3, and MTND4L | |||
| have been localized to the hydrophobic protein fragment (Ragan, 1987), and the MTND2, MTND4, and MTND5 | |||
| gene products probably reside there also. | |||
| The hydrophobic protein fragment contains the iron-sulfur centers that are the likely electron donors to | |||
| ubiquinone (Ohnishi et al., 1985; Ohnishi et al., 1974). | |||
| The 3644T-C mutation in ND1 converts amino acid 113, valine, to alanine. Munakata et al. (2004) noted that | |||
| val113 is well conserved from Drosophila to mammalian species | |||
| Complex III | CYTB | CYTB catalyzes the transfer of electrons from ubiquinol (reduced Coenzyme Q10) to cytochrome c and utilizes the | |
| energy to translocate protons from inside the mitochondrial inner membrane to outside. | |||
| These 11 complex III subunits include core proteins I and II, cytochrome b (subunit III), cytochrome c1 (subunit IV), | |||
| the Rieske iron-sulfur protein (subunit V), and several smaller polypeptides. | |||
| MTCYB is a highly evolutionarily conserved, hydrophobic protein containing 8 or 9 transmembrane domains | |||
| and 2 heme groups | |||
| The patient was found to have a 14849T-C mutation in the MTCYB gene, resulting in a substitution of a | |||
| �highly conserved serine for proline at position 35. | |||
| Complex IV | Complex IV is located within the mitochondrial inner membrane and is the third and final enzyme of the ETC | ||
| It collects electrons from reduced cytochrome c and transfers them to oxygen to give water. | |||
| Complex IV is composed of 13 polypeptides: 3mtEncoded and others nuclear DNA encoded | |||
| COX 1 | In mitochondrial Complex IV, the 2 hemes are a and a3 and the 2 coppers are CuA and CuB. The 2 hemes and CuB | ||
| are bound to subunit I. For mammalian MTCO1, there are 12 membrane-spanning alpha-helices (I to XII).� | |||
| Of these helices, heme a is located between helix II and X, ligated with the invariant histidines at amino acid | |||
| �102 (MTCO1 88) of helix II and at 421 (MTCO1 378) of helix X. Helix X lies between heme a and heme a3, | |||
| with heme a3 bound to the opposite side of helix X at invariant histidine at amino acid 419 (MTCO1 376). | |||
| �Heme a3 is a component of a binuclear center which includes CuB and where oxygen is reduced to water. | |||
| CuB is thought to lie adjacent to the iron of heme a3 and to be ligated to helix VI through invariant histidine | |||
| �284 (MTCO1 240) and to helix VII through invariant histidines 333 and 334 (MTCO1 290 and 291). | |||
| Amino acids histidine 411 (MTCO1 368), aspartate 412 (369), threonine 413 (370), and tyrosine 414 (371) | |||
| �occur in the conserved loop between helices IX and X, lying close to hemes a and a3, and may be in the proximity | |||
| �of the CuA located in subunit II (Hosler et al., 1993). | |||
| Jaksch et al. (1998) identified a G-to-A transition at nucleotide 6480 of the MTCO1 gene in a child, her mother,� | |||
| and sister with cytochrome c oxidase deficiency (220110) associated with sensorineural hearing loss, ataxia, | |||
| myoclonic epilepsy, and mental retardation. | |||
| COX 2 | It collects electrons from ferrocytochrome c (reduced cytochrome c) and transfers them to oxygen to give water. | ||
| Subunit II of Complex IV interacts with cytochrome c and contains the CuA center. This means that the pathway | |||
| �of electron transfer through Complex IV is from cytochrome c, to CuA, to cytochrome a, and then to the binuclear | |||
| � center of cytochrome a3-CuB. It is thought that the transfer of electrons from cytochrome a to the binuclear center | |||
| �is the key control point in the reaction and one of the major points of energy transduction (Hill, 1993). | |||
| CuA most likely resides in a loop containing conserved cysteines at amino acids 196 and 200 and a conserved | |||
| �histidine at 204, with the fourth ligand being histidine 161 | |||
| Cytochrome c interacts with subunit II through the association of a ring of lysines around the heme edge of | |||
| cytochrome c with carboxyls in subunit II, specifically glutamate 129, aspartate 132, and glutamate 198 | |||
| �(Hill, 1993; Capaldi, 1990). | |||
| The 25-np 3-prime-nontranslated sequence (5-prime-CACCCCCTCTACCCCCTCTAGAGGG) contains 2 9-np repeats | |||
| which are polymorphic in the world populations. One polymorphism involves the deletion of 1 repeat and is common | |||
| �in Asian, Polynesian and Native American mtDNAs. A second polymorphism involves additional Cs' inserted within | |||
| the runs of Cs (Cann and Wilson, 1983; Wrischnik et al., 1987; Hertzberg et al., 1989; Ballinger et al., 1992; | |||
| Schurr et al., 1990; Torroni et al., 1992; Torroni et al., 1993). | |||
| COX 3 | Subunit III is a highly conserved and ubiquitous subunit of Complex III, yet its function remains unclear | ||
| Cleavage at the 5-prime end occurs between the two As of the MTATP6 termination codon ACA TA //A UG ACC | |||
| �(ThrTerMetThr) creating transcript 15, the MTCO3 mRNA (Montoya et al., 1981; Ojala et al., 1981; | |||
| �Attardi et al., 1982). This is the only instance in which 2 separate transcripts, transcript 14� for MTATP8-MTATP6 | |||
| �and transcript 15 for MTCO3, are not separated by a tRNA. Hence, they must be processed by a separate system. | |||
| If the 14+15 transcript were not cleaved, then MTATP6 would be translated, but MTCO3 might not be translated | |||
| since its initiation codon overlaps the MTATP6 termination codon. Since the ratio between MTATP6 and MTCO3 | |||
| can vary between patient and control cell mitochondria, it is possible that the cleavage of transcript 14 + 15 | |||
| provides a mechanism for modulating the biogenesis of the electron transport chain relative to the ATP synthase | |||
| (Wallace et al., 1986). | |||
| Complex V | ATP6 | In an isolated case of mental retardation and ataxia without retinitis pigmentosa, de Coo et al. (1996) found an | |
| 8993T-G transversion (516060.0001). | |||
| Holt et al. (1990) found a heteroplasmic T-to-G transversion at nucleotide pair 8993 in a maternal pedigree which | |||
| resulted in the change of a hydrophobic leucine to a hydrophilic arginine at position 156 in subunit 6� of | |||
| mitochondrial H(+)-ATPase. The clinical symptoms varied in proportion to the percentage of mutant mtDNAs | |||
| �but the most common clinical presentation included neurogenic muscle weakness, ataxia, and retinitis | |||
| �pigmentosa, leading to the designation of NARP syndrome (551500). | |||
| �The insertion of an arginine in the hydrophobic sequence of ATPase 6 probably interferes with the hydrogen ion | |||
| �channel formed by subunits 6 and 9 of the ATPase, thus causing failure of ATP synthesis. | |||
| 8993T-G is a well known and established mutation for ataxia cases | |||
| ATP8 | |||
| tRNA | Transfer RNA | ||
| The default sites shown for each tRNA were scored because they were either reported to cause ataxia or lie at conserved | |||
| sites in the tRNA. The literature for each site is presented below. | |||
| tRNA-Ala | OMIM:590000 | G5650A#7 :� J. Med. Genet. 40: 752-757, 2003 | |
| tRNA-Arg | OMIM:590005 | X | |
| tRNA-Asn | OMIM:590010 | G5703A#5 : J. Clin. Invest. 92: 2906-2915, 1993 | |
| tRNA-Asp | OMIM:590015 | A7526G#5 : Am. J. Med. Genet. 137A: 170-175, 2005 | |
| tRNA-Cys | OMIM:590020 | X | |
| tRNA-Gln | OMIM:590030 | X | |
| tRNA-Glu | OMIM:590025 | T14709C#6 : Am. J. Hum. Genet. 56: 1026-1033, 1995 | |
| tRNA-Gly | OMIM:590035 | T9997C#8 : Am. J. Hum. Genet. 55: 437-446, 1994 | |
| T10010C#0 : Neurology 58: 1282-1285, 2002 | |||
| A10044G#8 : J. Biol. Chem. 278: 16828-16833, 2003 | |||
| tRNA-His | OMIM:590040 | G12192A#3 : . J. Hum. Genet. 67: 1617-1620, 2000 | |
| G12183A#7 : Neurology 60: 1200-1203, 2003 | |||
| G12147A#9 : Arch. Neurol. 61: 269-272, 2004; Neurology 62: 1420-1423, 2004 | |||
| tRNA-Ile | OMIM:590045 | A4317G#0 : J. Biol. Chem. 278: 16828-16833, 2003 | |
| A4269G#0: Biochem. Biophys. Res. Commun. 186: 47-53, 1992 | |||
| G4284A#5 : Ann. Neurol. 51: 118-122, 2002 | |||
| G4300A#3 : J. Am. Coll. Cardiol. 41: 1786-1796, 2003 | |||
| tRNA-Leu | OMIM:590050 | A3243G#4 : Nature 348: 651-653, 1990; Biochem. Biophys. Res. Commun. 173: 816-822, 1990 | |
| T3271C#4 : Biochim. Biophys. Acta 1097: 238-240, 1991 | |||
| C3256T#3 : J. Clin. Invest. 92: 2906-2915, 1993 | |||
| C3303T#5 : Hum. Mutat. 3: 37-43, 1994 | |||
| T3252C#2 : Hum. Molec. Genet. 2: 2081-2087, 1993 | |||
| A3251G#3 : Quart. J. Med. 86: 709-713, 1993 | |||
| A3260G#1 :� J. Clin. Invest. 93: 1102-1107, 1994 | |||
| T3250C#3 : J. Pediat. 130: 138-145, 1997 | |||
| T3290C#2 : Acta Paediat. 88: 957-960, 1999 | |||
| A3274G#1 : Neurology 57: 1930-1931, 2001 (not in white blood cells ) | |||
| G3249A#1 : Arch. Neurol. 58: 1113-1118, 2001 | |||
| tRNA-Leu2 | OMIM:590055 | A12320G#5 : Am. J. Hum. Genet. 60: 373-380, 1997 (mutation present only in skeletal muscle) | |
| T12297C#6 : Europ. J. Hum. Genet. 9: 311-315, 2001 | |||
| tRNA-Lys | OMIM:590060 | A8344G#9 : Muscle Nerve 17: 52-57, 1994 (This is one of the reference that associates this site with ataxia) | |
| T8356C#7 : Am. J. Hum. Genet. 51: 1213-1217, 1992 | |||
| G8363A#6 : Am. J. Hum. Genet. 58: 933-939, 1996 | |||
| G8313A#7 : Pediat. Res. 42: 448-454, 1997 | |||
| G8361A#3 : Biochem. Biophys. Res. Commun. 245: 523-527, 1998 | |||
| tRNA-Met | OMIM:590065 | X | |
| tRNA-Phe | OMIM:590070 | G583A#8 : J. Neurol. Neurosurg. Psychiat. 65: 512-517, 1998 | |
| tRNA-Pro | OMIM:590075 | G15990A#3 : Nature Genet. 4: 284-288, 1993 (not detected in WBC) | |
| T15965C#1 : Neurogenetics 2: 121-127, 1999 (histologically proven idiopathic Parkinson disease ) | |||
| tRNA-Ser | OMIM:590080 | T7512C#6 : Biochem. Biophys. Res. Commun. 214: 86-93, 1995; J. Med. Genet. 35: 895-900, 1998 | |
| A7445G#8 : Molec. Cell. Biol. 18: 5868-5879, 1998 | |||
| X7472C#3 : Hum. Molec. Genet. 4: 1421-1427, 1995; J. Med. Genet. 35: 895-900, 1998 | |||
| T7510C#5 : J. Med. Genet. 37: 692-694, 2000 | |||
| tRNA-Ser2 | OMIM:590085 | C12258A#7 : Am. J. Hum. Genet. 64: 971-985, 1999 | |
| tRNA-Thr | OMIM:590090 | G15950A#1 : Neurogenetics 2: 121-127, 1999 | |
| tRNA-Trp | OMIM:590095 | G5549A#7 : Ann. Neurol. 37: 400-403, 1995 | |
| X5537T#4 : Neuropediatrics 34: 87-91, 2003; Ann. Neurol. 42: 256-260, 1997 | |||
| G5521A#2 : Neuromusc. Disord. 8: 291-295, 1998 (not detected in leukocytes) | |||
| tRNA-Tyr | OMIM:590100 | A5874G#4 : Neurology 55: 1210-1212, 2000 | |
| X5885T#6 : Neurology 57: 2298-2301, 2001 | |||
| G5877A#5 : J. Med. Genet. 38: 703-705, 2001 | |||
| A5843G#5 : Am. J. Med. Genet. 123A: 172-178, 2003 | |||
| tRNA-Val | OMIM:590105 | G1606A#9 : Ann. Neurol. 43: 98-101, 1998; . Neurol. 59: 1013-1015, 2002 | |
| C1624T#7 : Nature Genet. 30: 145-146, 2002 | |||